Spinal Muscular Atrophy (SMA)
Skeletal Muscle and Neuromuscular Junction (NMJ) Components
Identifying diverse modalities including small molecules and regenerative progenitor cells to restore functional skeletal muscle and neuromuscular junction in SMA
Learn more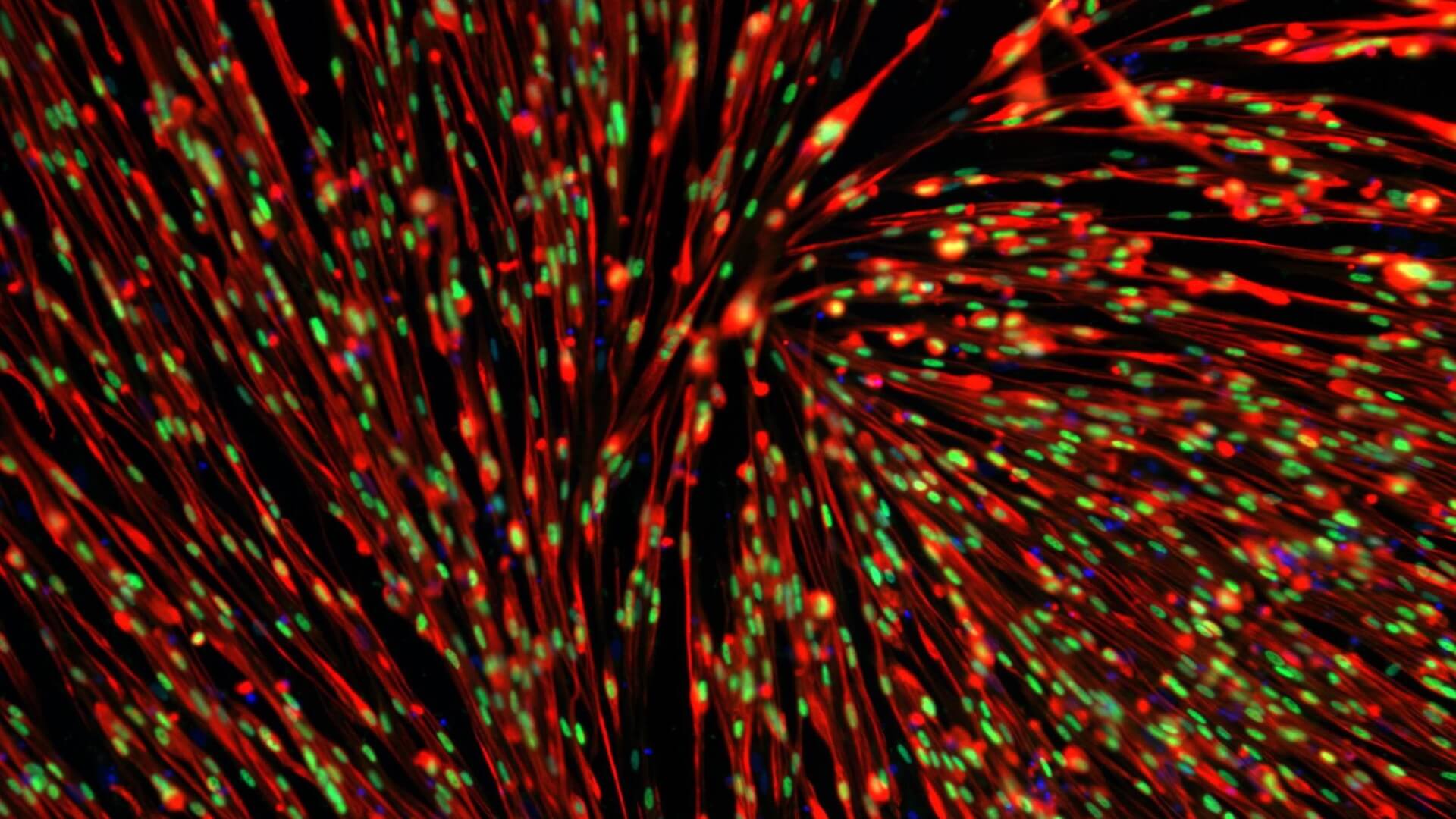
Prevalence/ Incidence
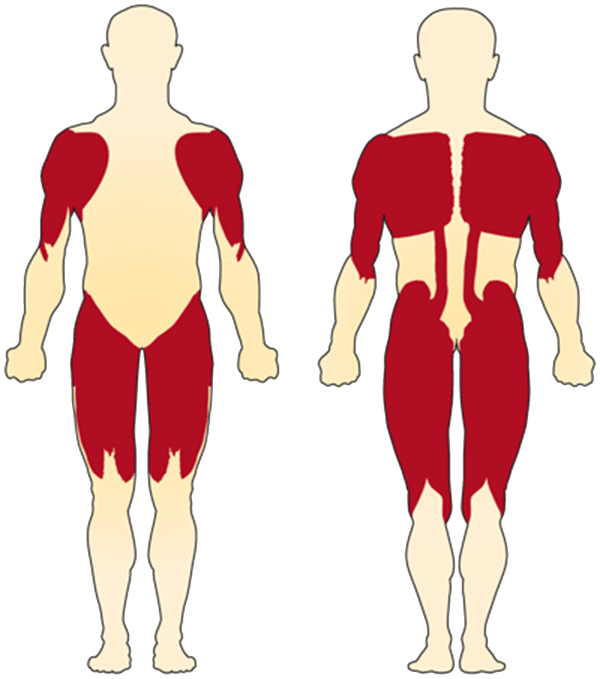
- Muscle atrophy and loss of motor neurons in the anterior horn of the spinal chord to yield muscular weakness
- Proximal muscles are most severely affected
- Muscle weakness is the most common symptom
- Cognitive and emotional development are unaffected
- Orphan drug designation by the FDA
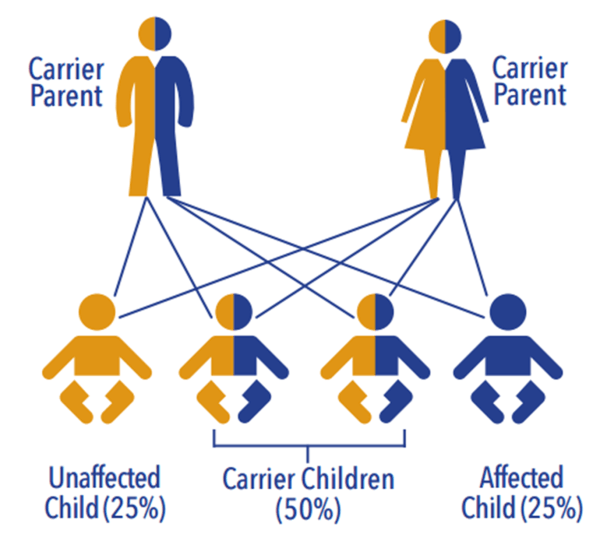
- After CF, the most common autosomal recessive inherited disorder
- SMA was granted orphan drug designation by the FDA
- 1 in 40 people are carriers
- 1 in 6,000-10,000 children born with SMA
- Estimated number of patients in US is ~9,000
SMA is Caused by Defects in the SMN1 gene
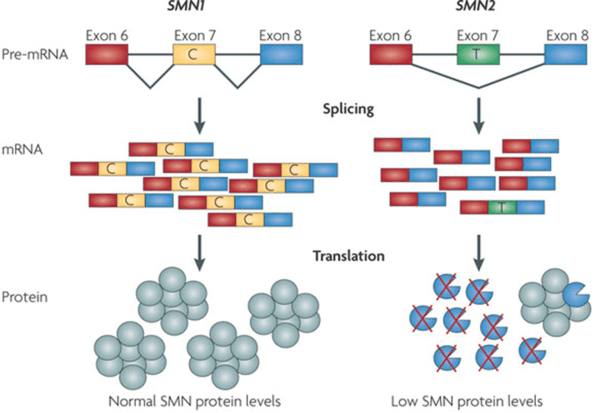
Associated with mutations in the survival of motor neuron gene (SMN1)
- 2 forms of SMN gene on each allele: a telomeric (SMN1) and centromeric form (SMN2)
- Transcription of the SMN1 gene produces full-length messenger RNA that encode the SMN protein
- The SMN2 gene is identical to the SMN1 gene except for a C to T substitution at position 840 to exclude exon 7 during transcription.
- The resultant truncated protein is not function and rapidly degrades.
- The exclusion of exon 7 is not complete, a small fraction of total mRNA transcript (~10-20%) encodes full-length, functional SMN
- Copy number of SMN2 in SMA patients ranges from 1-5, there is a positive correlation between SMN2 copy number and milder phenotypes
SMA Varies in Severity
SMA has a continuous spectrum of symptoms that ranges from very severe to mild across the four classifications of SMA types
| Type | Severity | Age of onset | Life expectancy | Characteristics |
|---|---|---|---|---|
| I (Werdnig-Hoffman disease) |
Severe | 0-6 months | <2 years | Quick and unexpected onset (“floppy baby syndrome”). Rapid motor neuron death causes respiratory complications that are frequent cause of death. Unless placed on mechanical ventilation, SMA type 1 patients do not live past 2 years. |
| II | Intermediate | 7-18 months | >2 years | Child is never able to stand and walk but are able to maintain a sitting position at some point. Weakness usually observed between 6-18 months. Muscle weakness is progressive and respiratory system is a major concern. |
| III (Kugelberg-Welander disease) |
Mild | >18 months | Adult | Juvenile form that usually manifests after 1 year of age. Patients are able to walk without support at some time, by may loss this ability. Respiratory involvement is less noticeable and life expectancy is normal or near normal. |
| IV (adult form) |
Mildest | Second and third decade | Adult | Adult-onset form usually manifests in the third decade of life with gradual weakening of the muscles. Life expectancy is normal. |
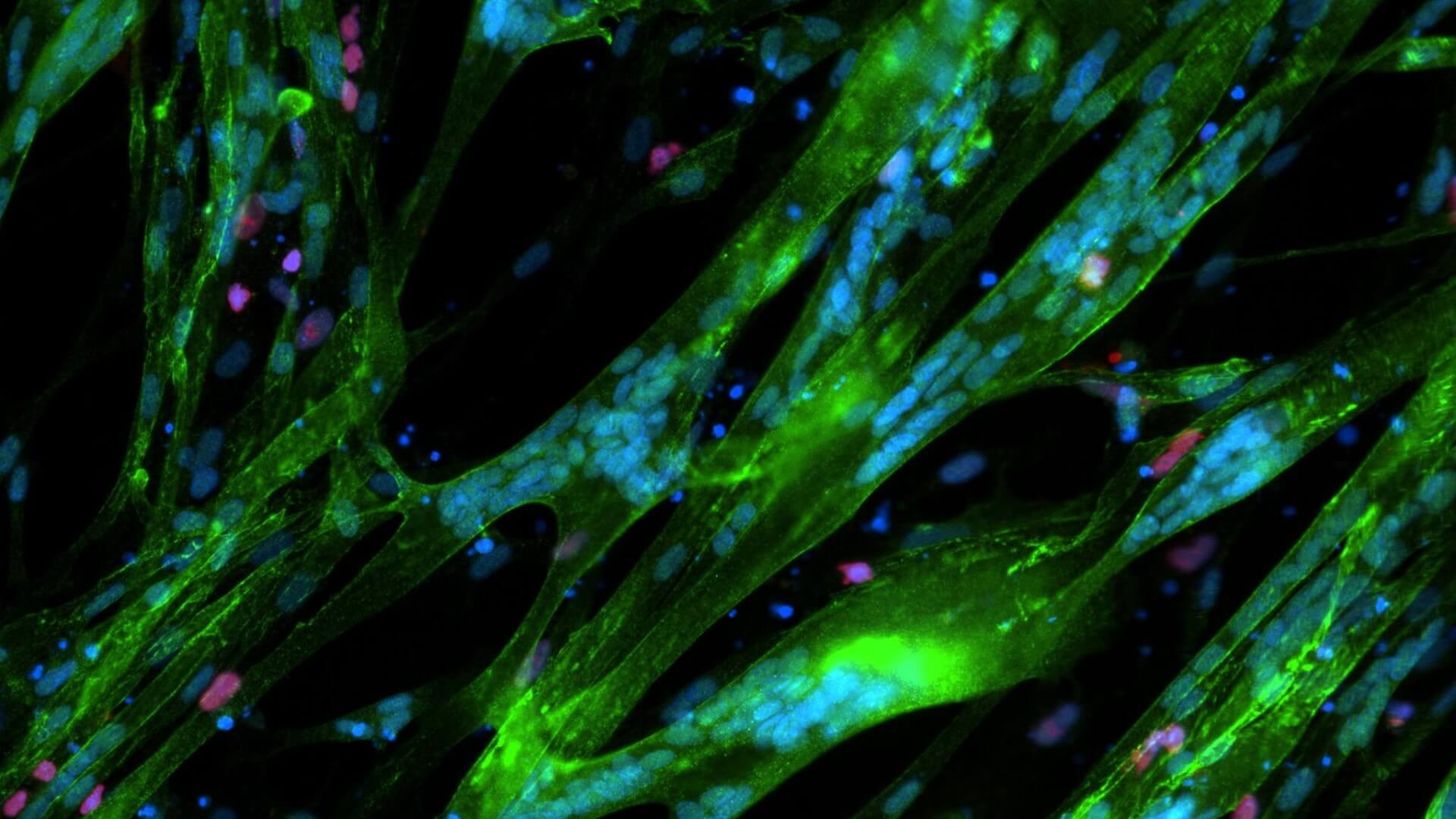
Developmental delay in SMA muscle
MYOCEA: ‘primary screening edge’



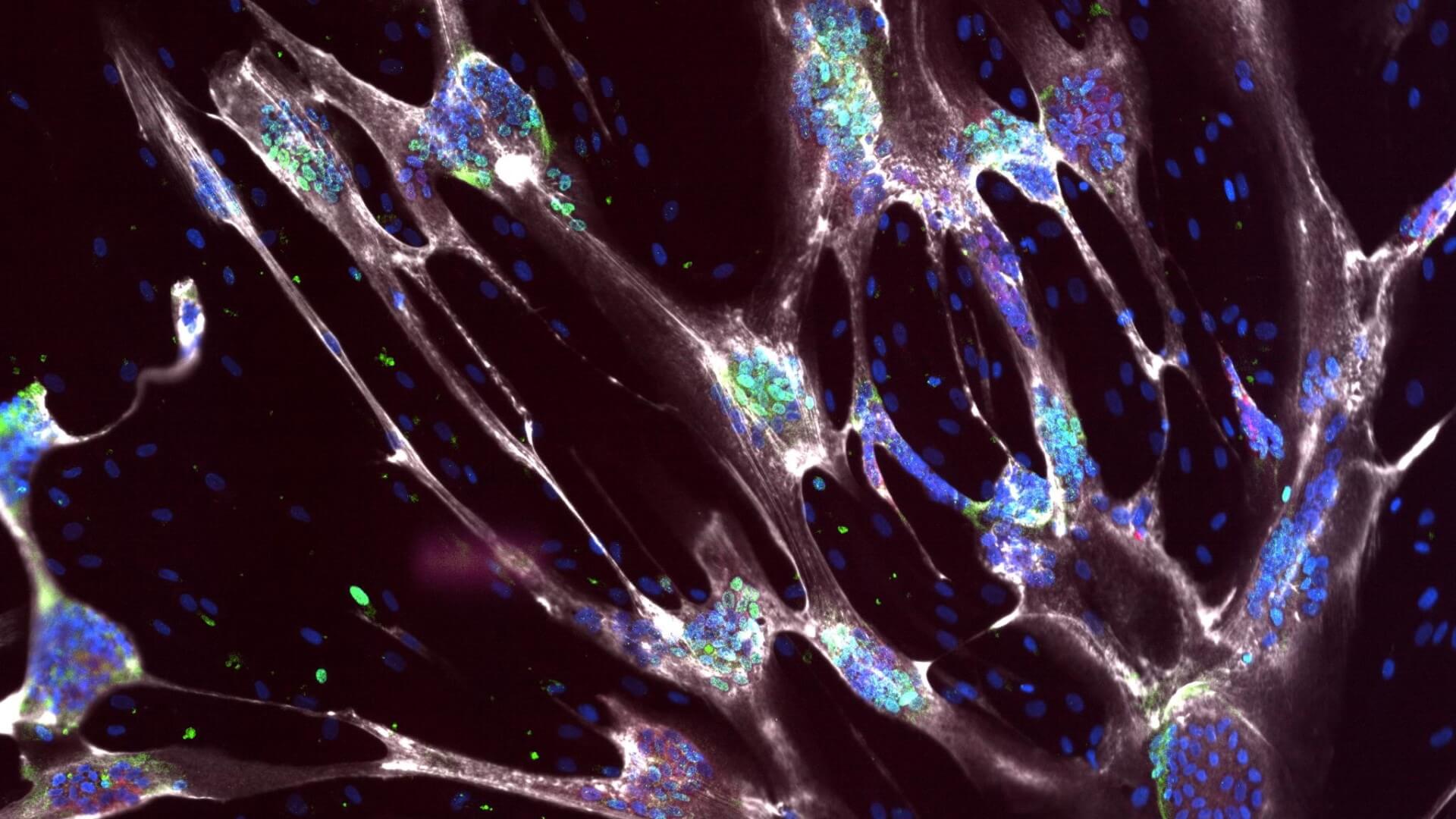
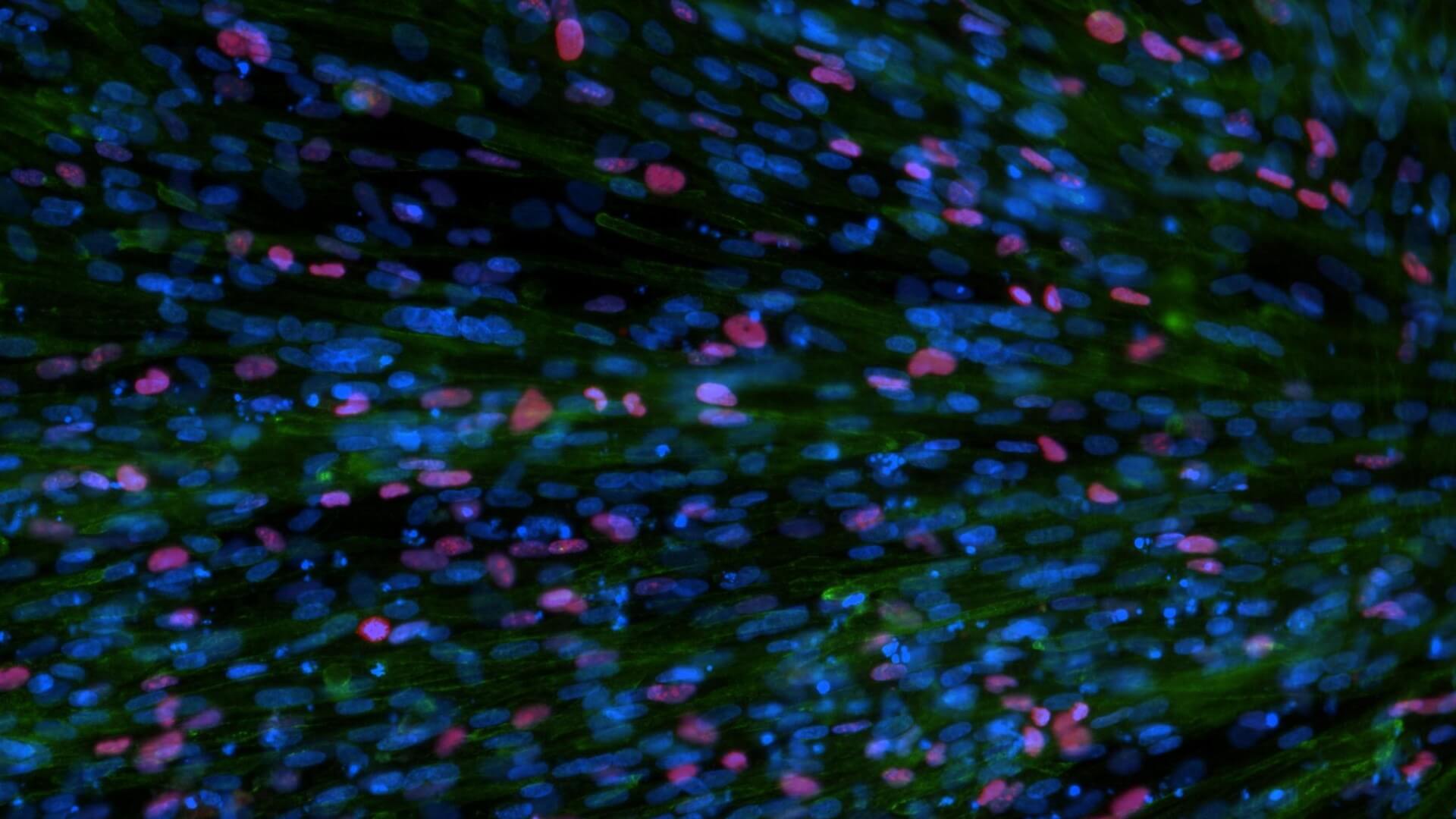
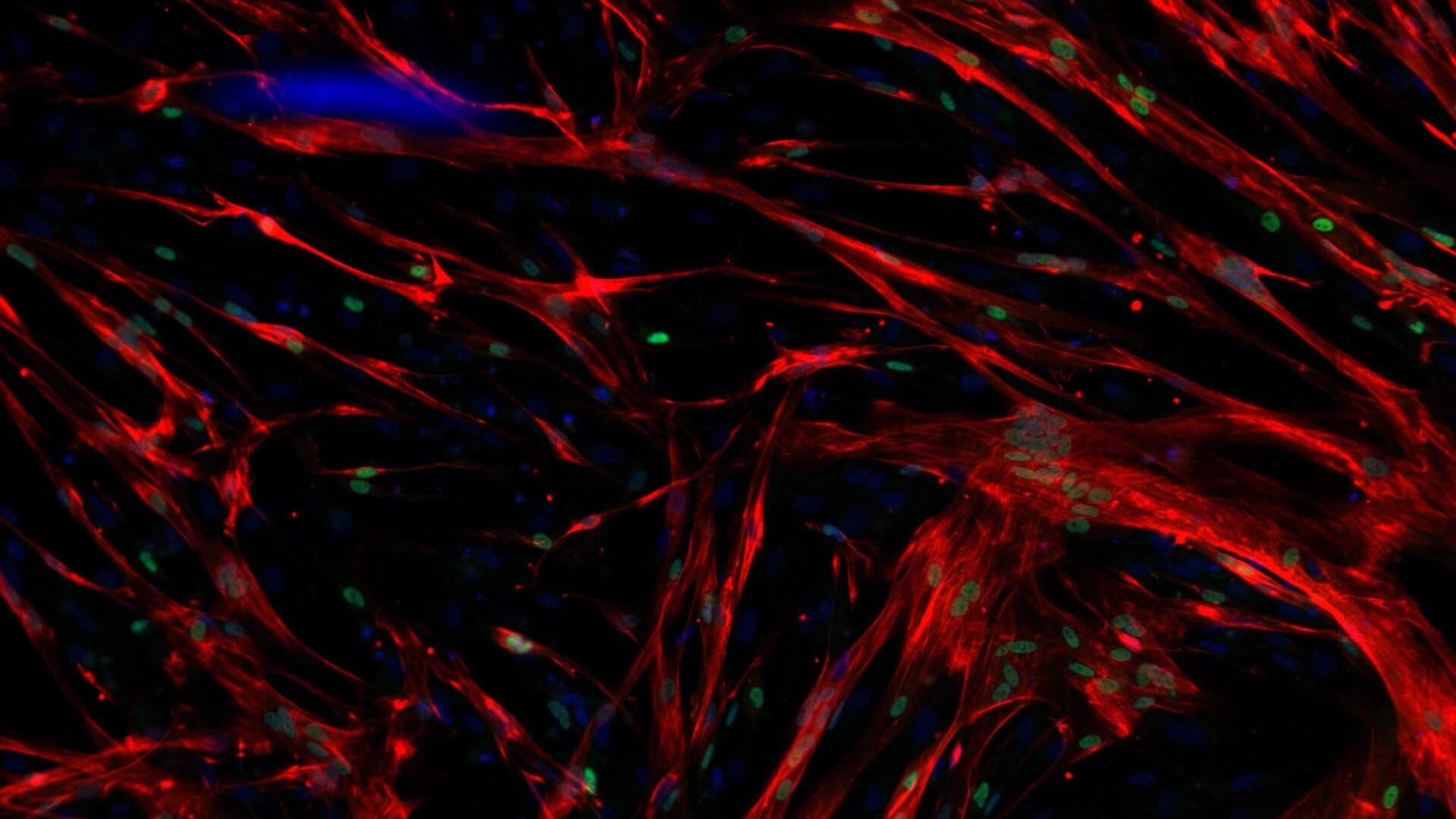
hPSC model of SMA: developmental delays in myoblast differentiation
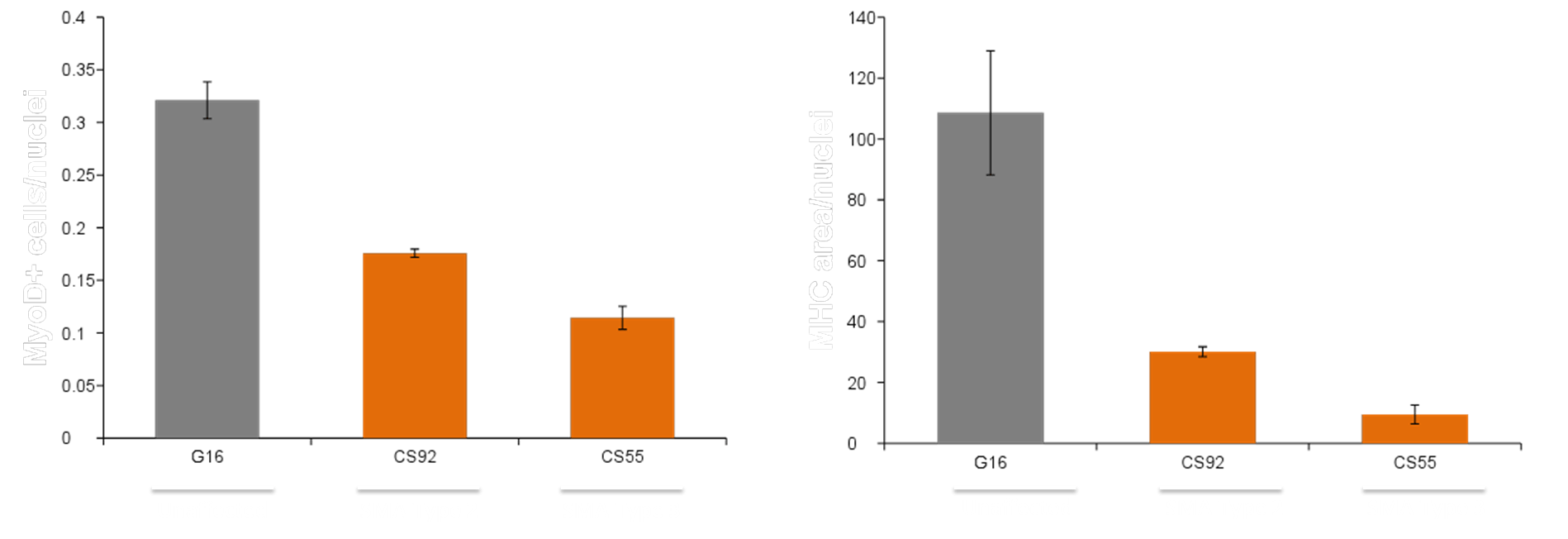
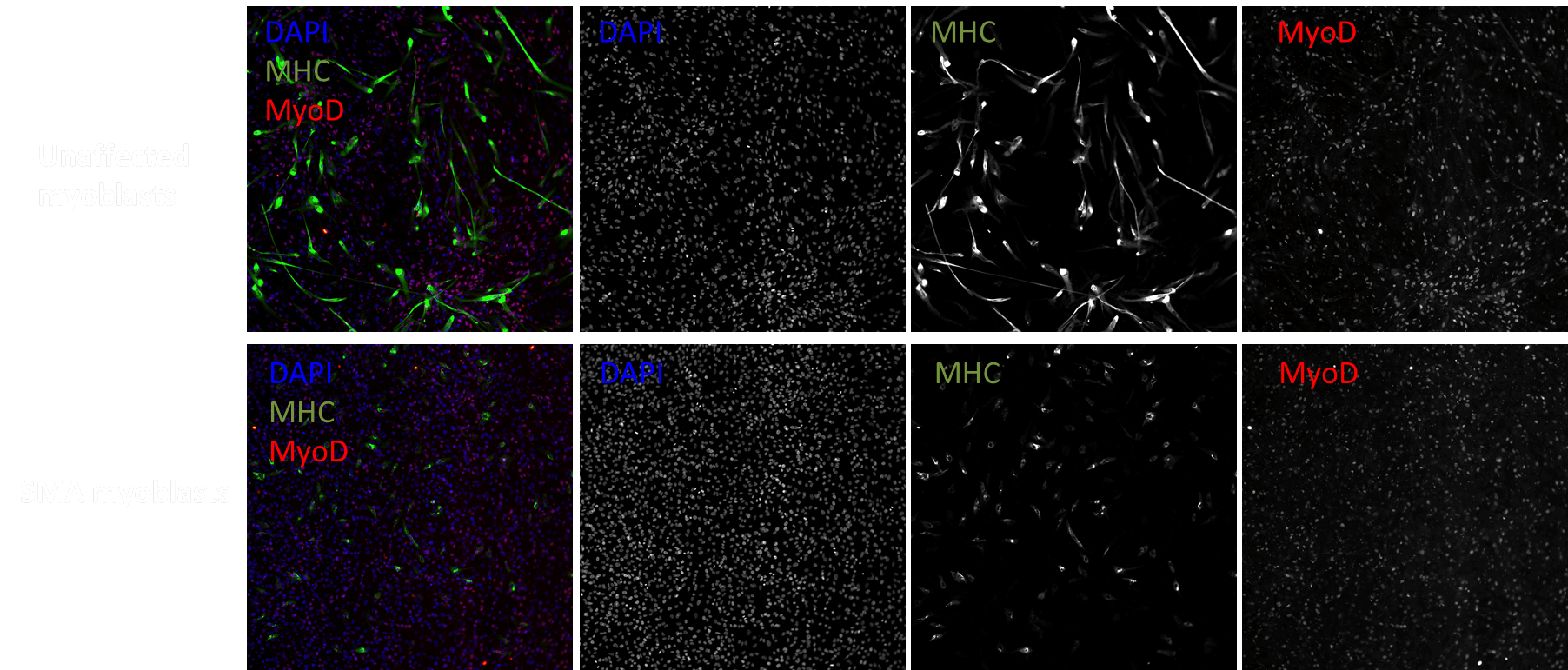 SMA myoblasts show reductions in MHC expression and the number of MyoD+ cells
SMA myoblasts show reductions in MHC expression and the number of MyoD+ cells
Developmental Delay: Quantitation of MHC and MyoD expression in
SMA vs. unaffected myoblasts

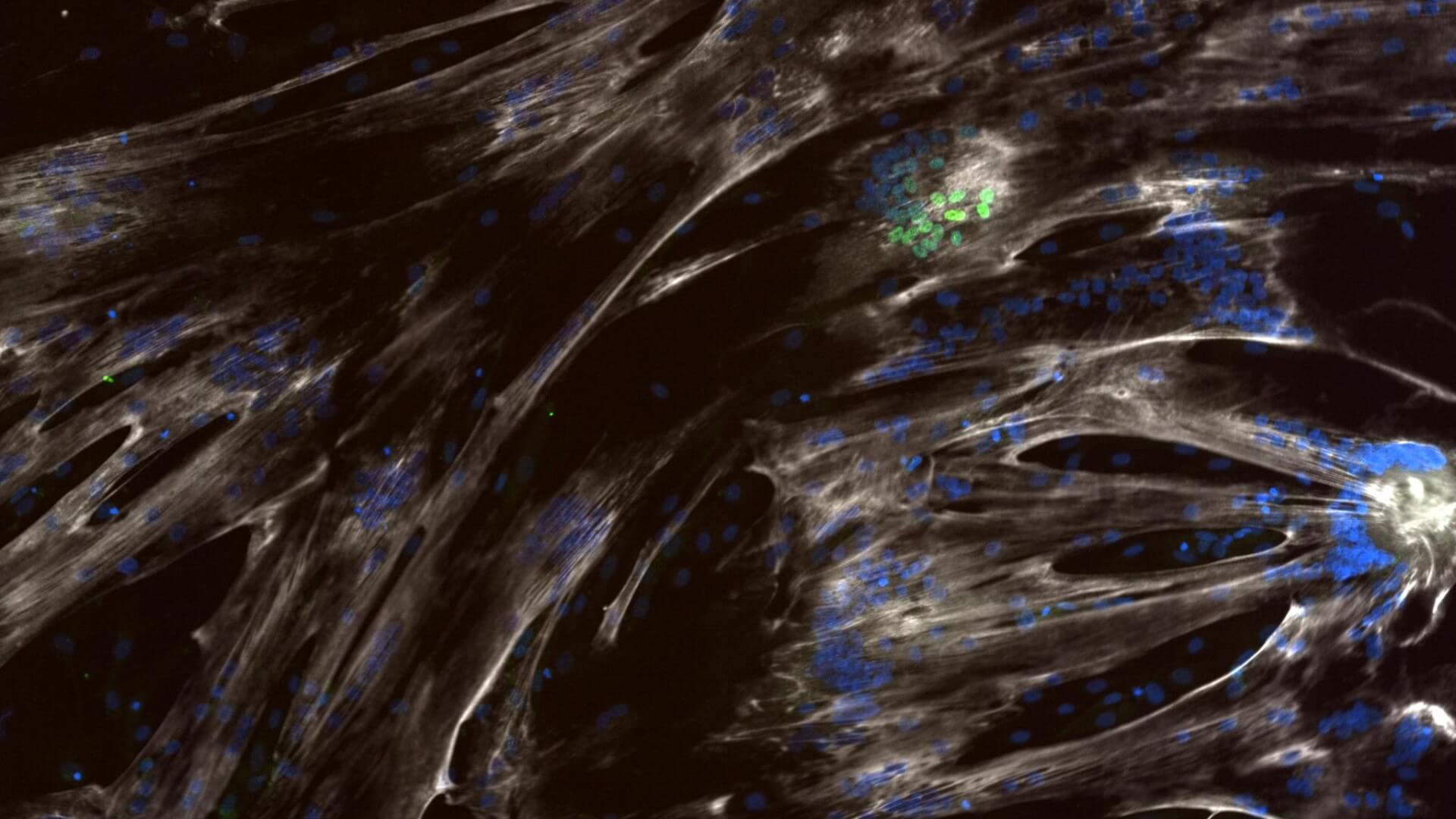
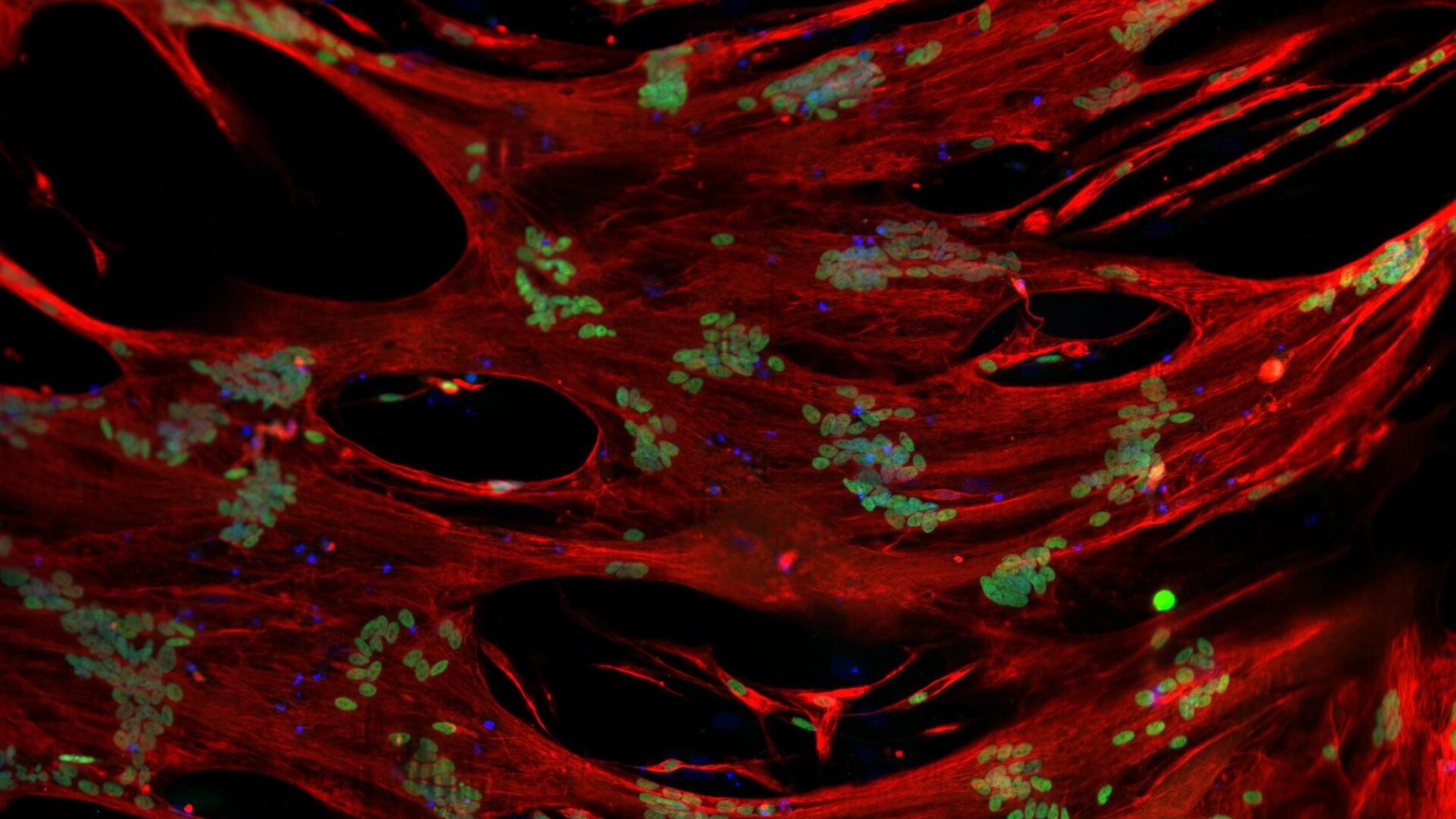
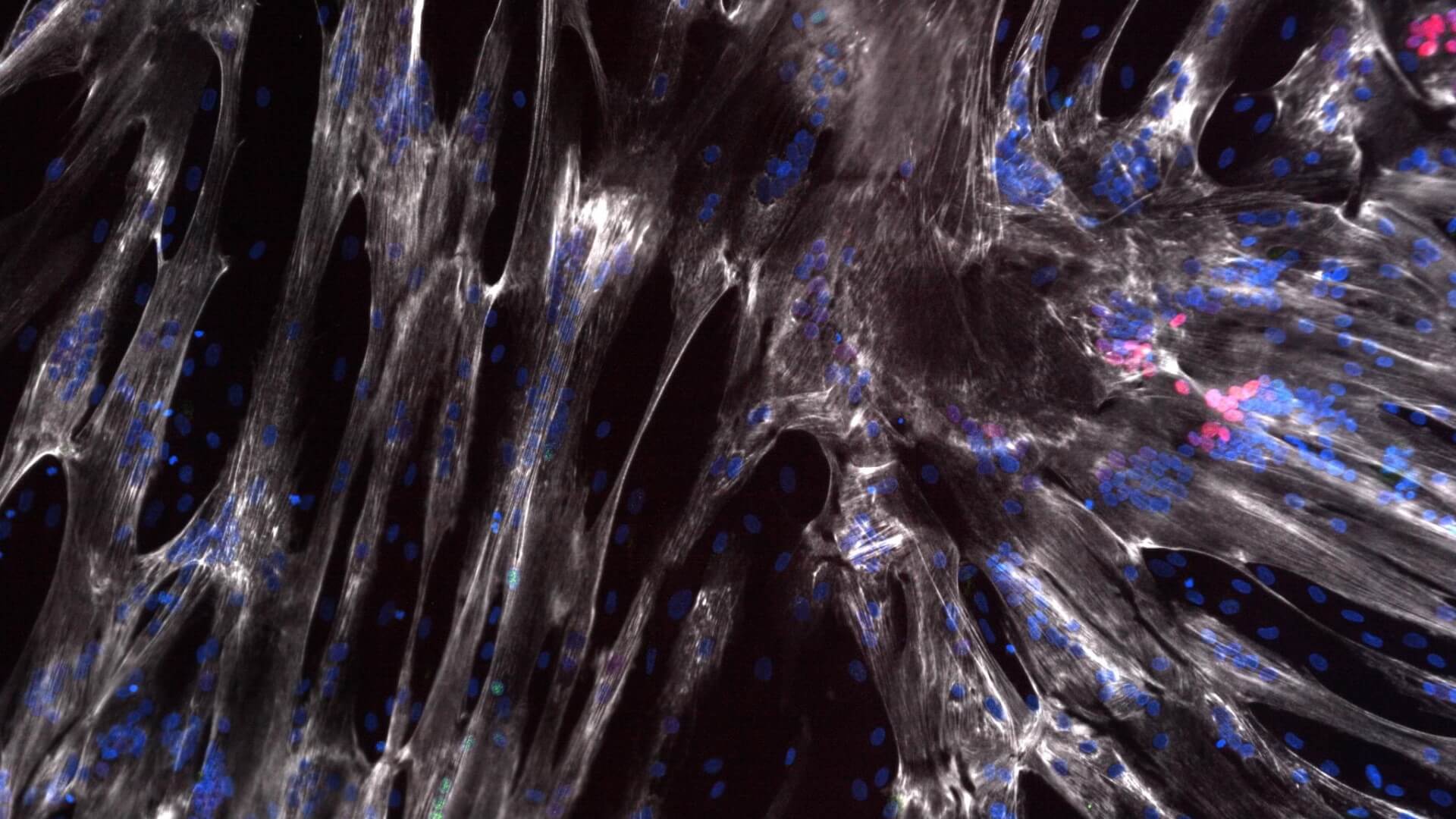
Developmental delay in iPSC-derived myoblasts from SMA Type 1 lines

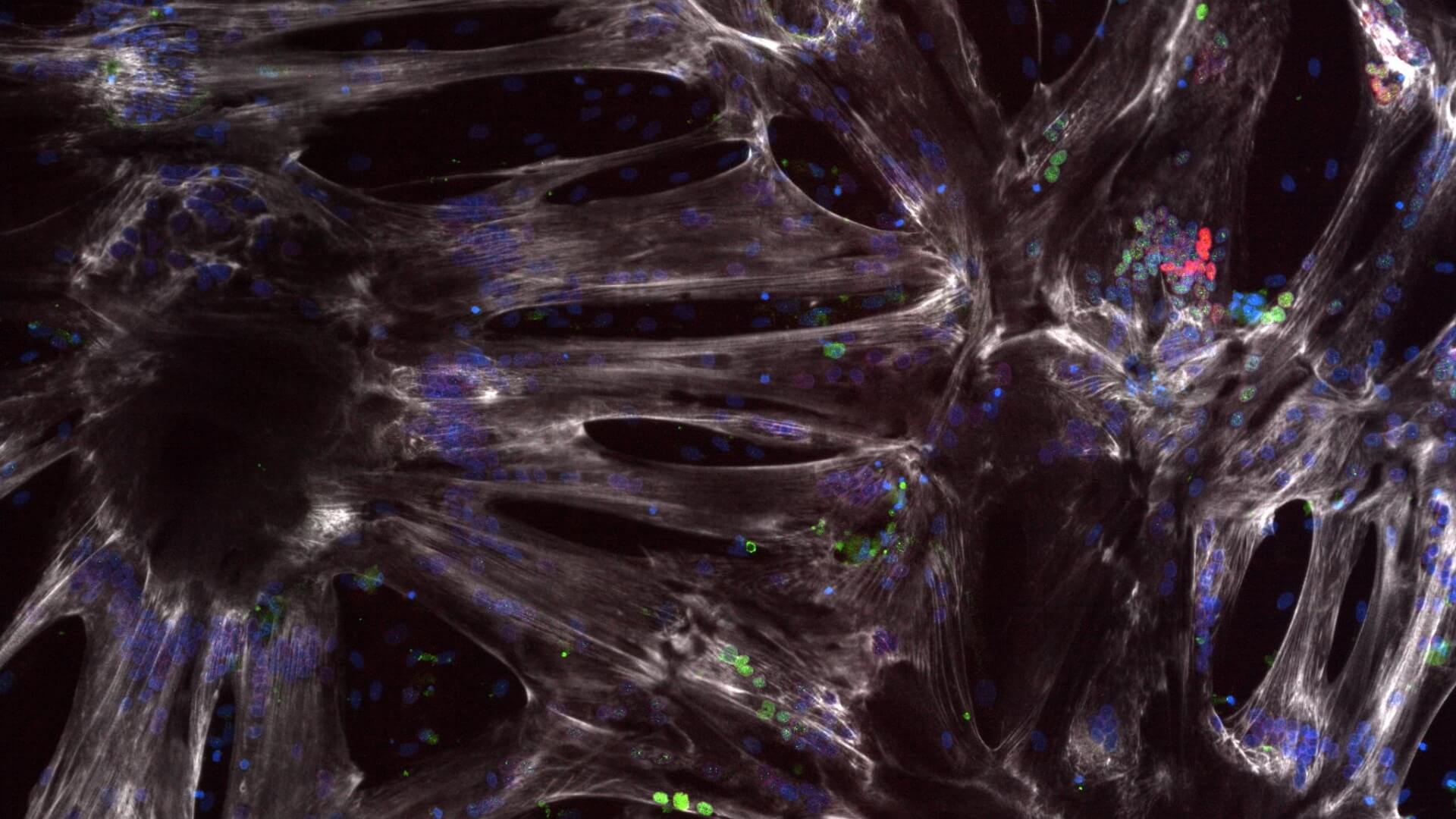
SMA Type 2 and 3 iPSC lines show a similar developmental delay

SMA Type 2 and 3 iPSC lines show a similar developmental delay
 SMA Type 2 and 3 show a developmental delay phenotype
SMA Type 2 and 3 show a developmental delay phenotype
MYOCEA: Summary of EC50s for top compounds
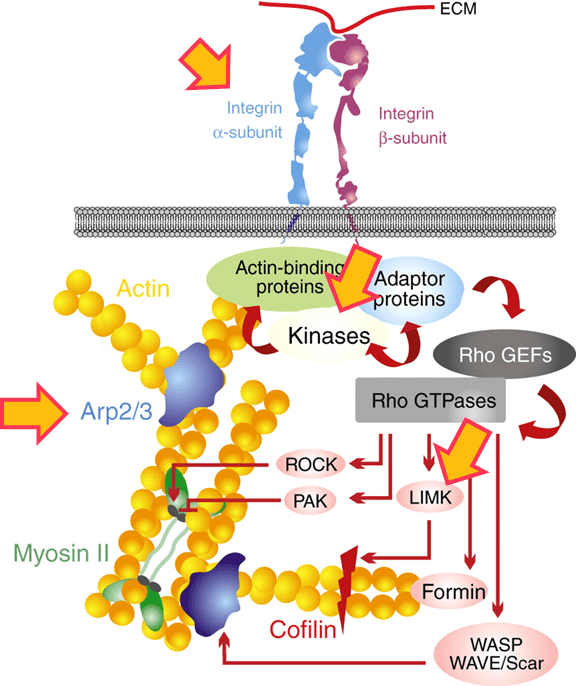
Signaling Networks Modulated
| Compound | Pathway | MYOCEA102 MHC EC50 |
MYOCEA102 MyoD EC50 |
CS32iSMA1 MHC EC50 |
CS32iSMA1 MyoD EC50 |
|---|---|---|---|---|---|
| MYO1 | Network 1 | 15 nM | 6 nM | 7 nM | 3 nM |
| MYO2 | Network 2 | 13 nM | 6 nM | 640 nM | <1 nM |
| MYO3 | Network 3 | 55 nM | 1.2 uM | ||
| MYO4 | Integrin inhibitor | 120 nM | 770 nM | 4 uM | |
| MYO5 | Actin polymerization inhibitor |
10 nM | 950 nM | ||
| MYO6 | Src inhibitor | 40 nM | 120 nM | ||
| MYO7 | LIM kinase and ROCK inhibitor |
310 nM | 1.1 uM | ||
| MYO8 | Network 4 | 110 nM | 40 nM | 100 nM | 250 nM |
| MYO9 | Network 5 | 30 nM | 740 nM |
MYOCEA: SMA Secondary Assays
Capitalizing on reported clinical pathologies
| Published SMA phenotype | Secondary assay |
|---|---|
| Decreased muscle function | Nuclei/myofiber |
| Fusion index | |
| Myotube size/diameter | |
| Autophagy | Activation of Ubiquitin pathway |
| Expression of p62, beclin, LC3 | |
| RFP-GFP-LC3B Autophagy Sensor | |
| Mitochondrial function | Bioenergetics: Seahorse mito stress test |
| Mitochondrial DNA content | |
| Mitochondrial membrane polarization | |
| Mitochondrial complex protein expression | |
| Neuromuscular Junction | NMJ co-culture |
| Agrin induced AcH reorganization | |
| Motor neuron function |
MYOCEA: Hits rescue bioenergetic phenotype in SMA myoblasts
Pharmacological response (100 nM and 1 uM)
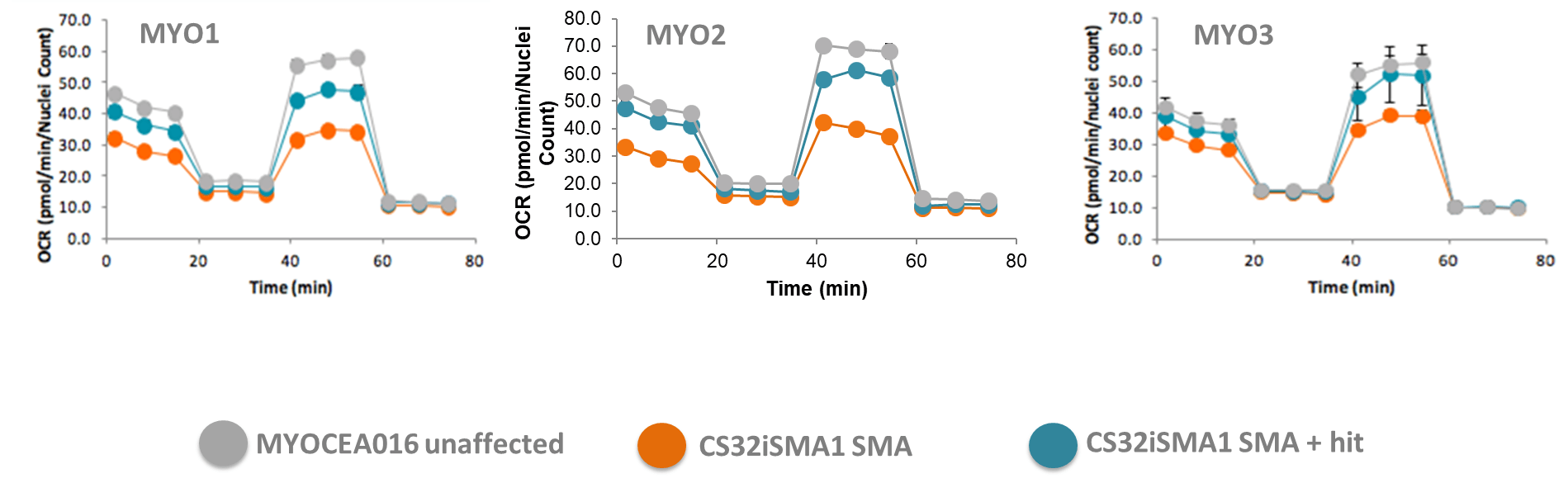
Secondary assays: SMA myoblasts have a mitochondrial membrane polarization defect
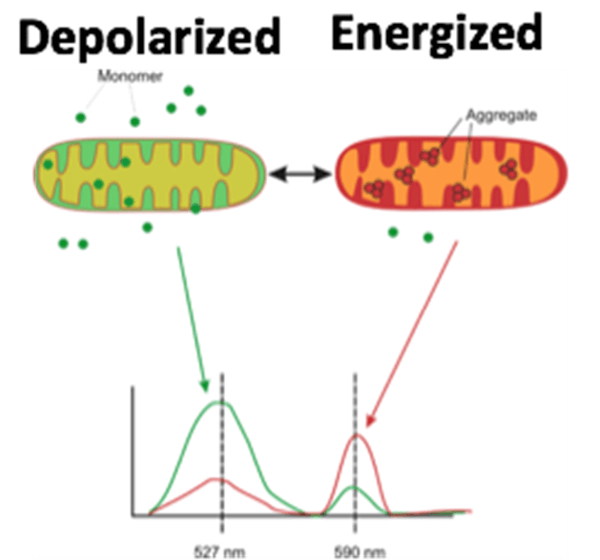 Fig1. JC-10:
Fig1. JC-10:
‧ Much more soluble than JC-1
‧ Monomer: green = depolarized mitochondria
‧ Trimer: red = intact, polarized mitochondria
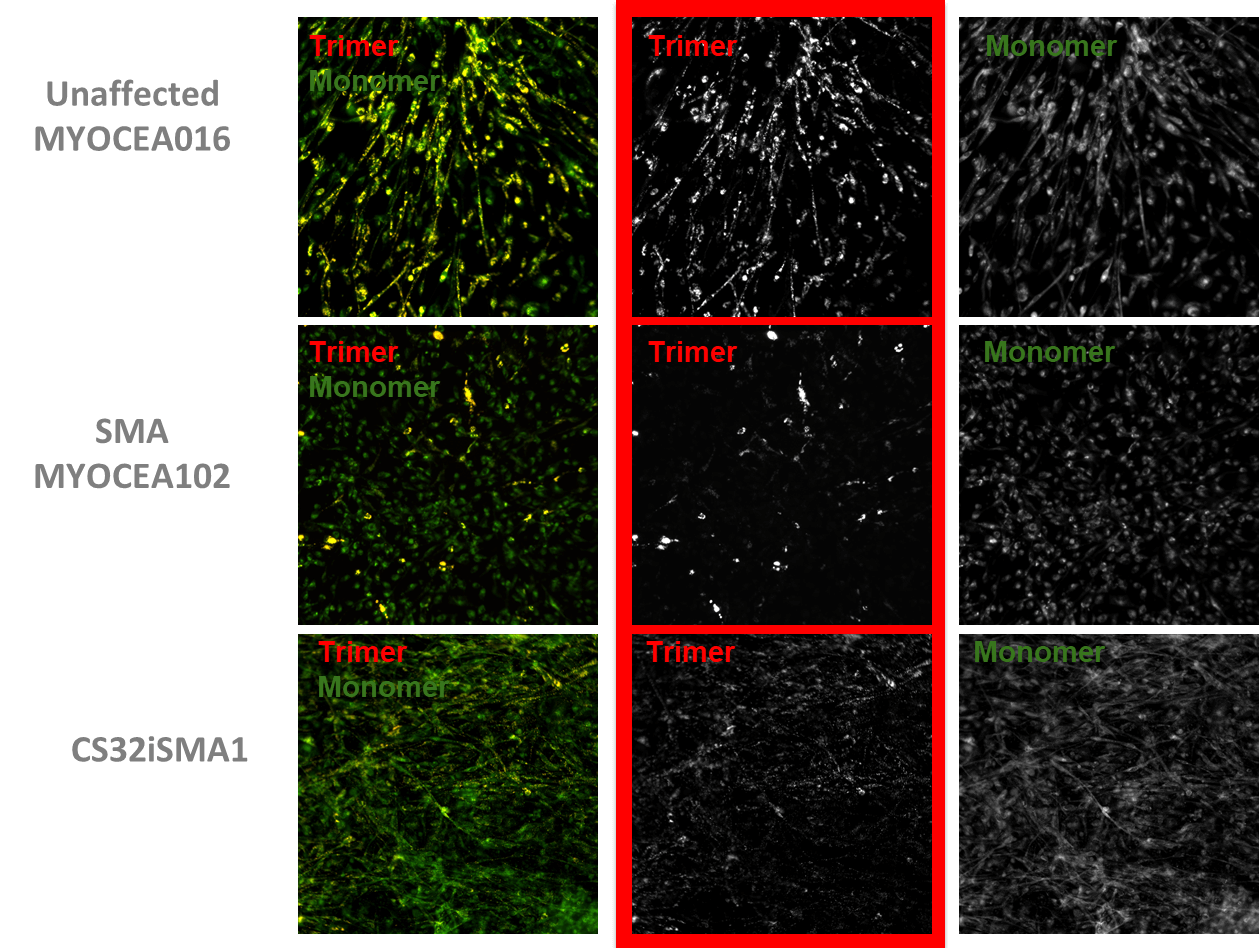
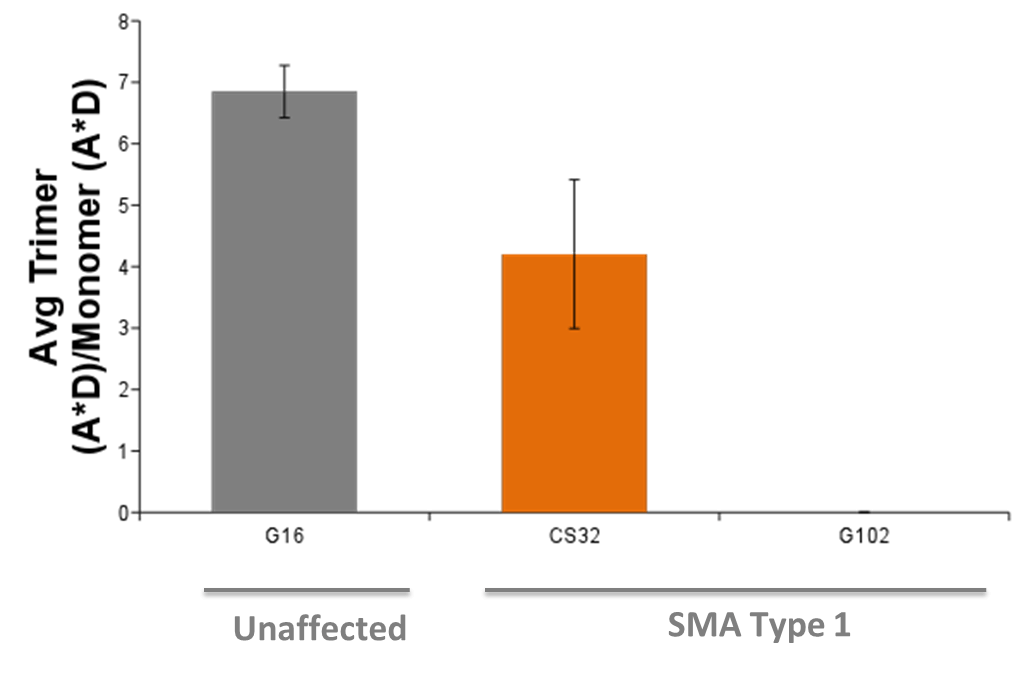 Fig2. All SMA lines tested show mitochondrial hypopolarization in their resting state
Fig2. All SMA lines tested show mitochondrial hypopolarization in their resting state
JC-10 staining of mito membrane polarization in myoblasts
MYOCEA: SMA, Nanostring™ Assay
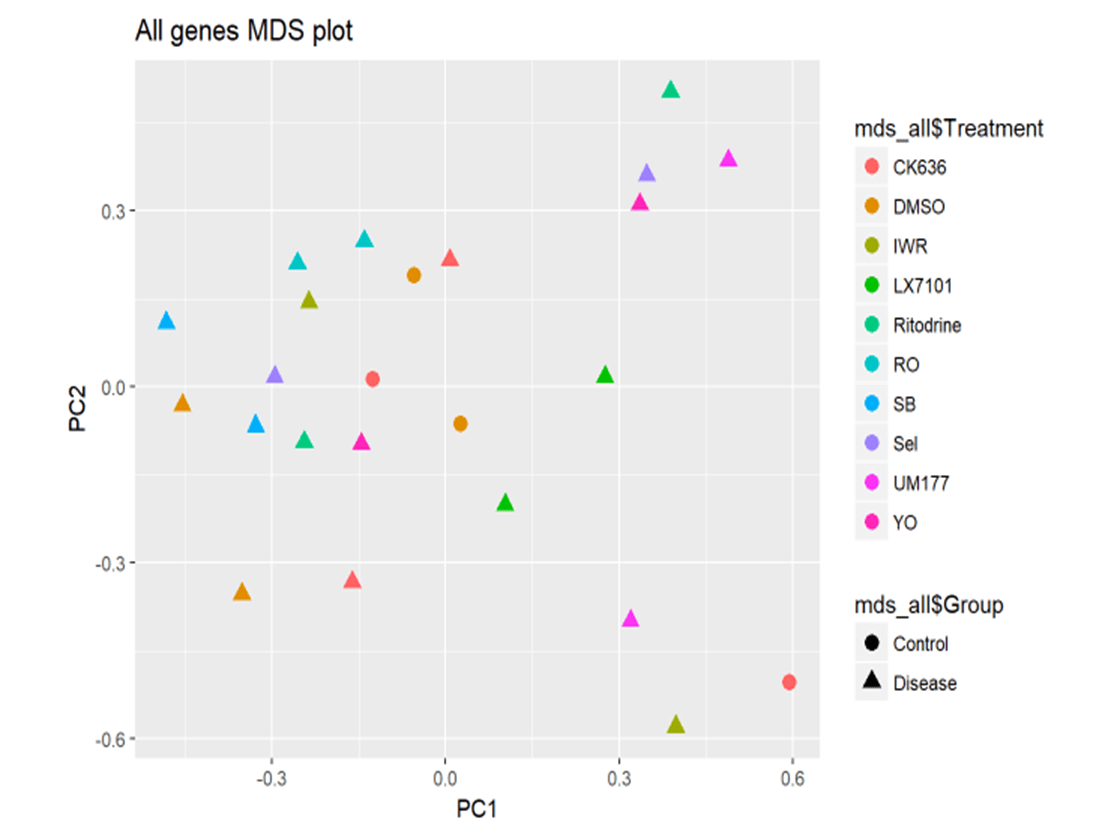
Treatment of SMA myoblasts with hits brings gene expression profile closer to unaffected cells

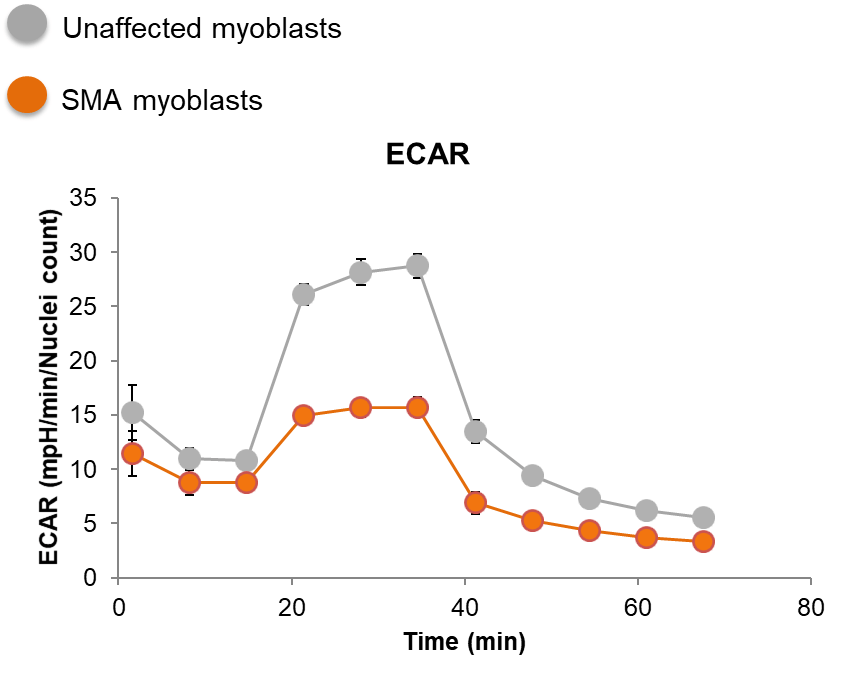
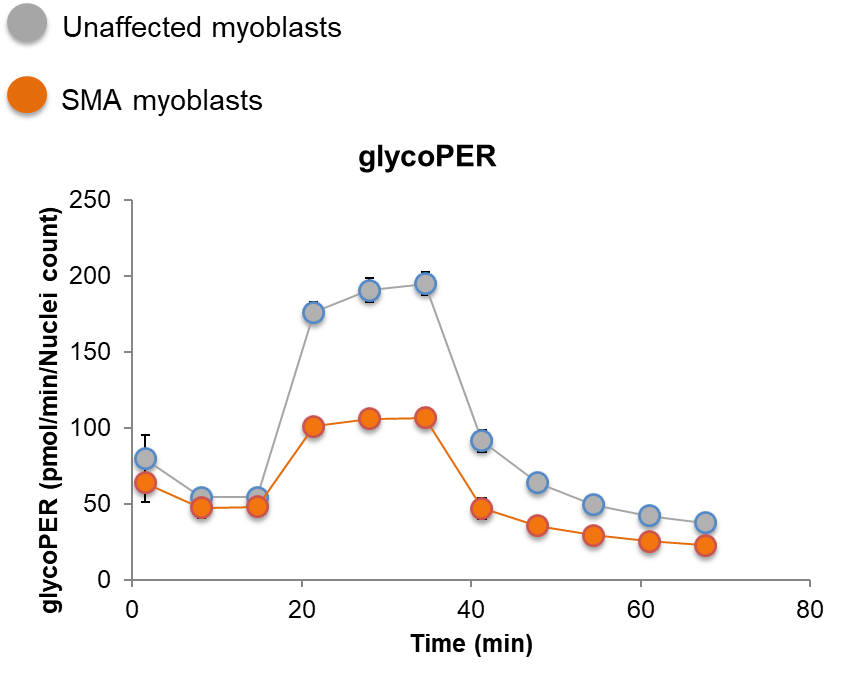
SMA myoblasts have a glycolytic deficit
Contact Us
Want to say hello? Want to know more about us? Give us a call or drop us an email and we will get back to you as soon as we can.